
페닐케톤뇨증(Phenylketonuria, PKU)은 아기가 태어날 때 선천적으로 갖는 희귀 유전대사질환이다. 페닐케톤뇨증은 페닐알라닌(phenylalanine, Phe)을 티로신으로 전환하는 페닐알라닌 수산화효소(phenylalanine hydroxylase, PAH)의 결핍으로 인해 페닐알라닌이 체내에 축적돼 발병한다.
페닐케톤뇨증을 가지고 태어난 아기는 페닐알라닌을 분해할 수 없다. 태어나자마자 혈류에 페닐알라닌 수치가 높아지기 시작한다. 페닐알라닌 과다 축적이나 섭취는 아기의 뇌에 독성을 미친다. 치료하지 않으면 뇌 손상을 유발할 수 있으며, 발달지연 및 지적장애와 같은 심각한 합병증으로 이어질 수 있다.
다행히 미국과 한국 등 주요 국가에서는 병원에서 태어나는 모든 아기는 출생 직후 필수 선별검사로 PKU 검사를 받는다. 담당 의사가 PKU로 진단하면 즉시 치료를 시작할 수 있다. 따라서 조기치료로 심각한 증상을 막고 정상적으로 발달할 가능성이 높다.
유병률과 증상
전세계 유병률은 2만3930명당 1명꼴로 45만명의 환자가 이 질환을 갖고 있는 것으로 추산된다. 지역별 편차가 커서 튀르키예(6667명당 1명), 이탈리아나 아일랜드 등에서는 2700명~4500명당 1명꼴로 높다. 중동 지역도 높은 편이다. 같은 아시아 지역에서도 중국은 1만5924명당 1명꼴로 상대적으로 높지만 태국은 22만7273명당 1명꼴로 매우 낮다. 전반적으로 아시아는 유럽이나 중동에 비해 유병률이 낮다. 유럽에서도 핀란드는 10만명당 1명꼴로 적은 편에 속한다. 미국에서 아프리카계(흑인)는 5만명당 1명꼴로 낮은 편이다. 아프리카 자체는 통계가 없다.
PKU 아기는 출생 시 건강해 보인다. 하지만 치료를 받지 않으면 PKU 증상은 3개월에서 6개월에 걸쳐 서서히 나타납니다. 신체적 증상은 메스꺼움부터 떨림까지 다양하다. 대략 1세가 되면 △숨, 땀, 소변의 곰팡이 냄새 △피부, 머리카락, 눈의 변화(가족의 다른 구성원에 비해 색이 밝음) △습진 △성장 지연 △메스꺼움과 구토 △떨림 △발달지연 및 지적장애 등이 나타날 수 있다.
이는 잦은 짜증과 자해 등 행동 문제로도 이어진다. 예컨대 과잉행동, 학습장애, 발작 등이 초래된다.
임신부가 PKU를 치료하지 않고 임신하면 유산할 위험이 높다. 관리되지 않는 PKU 질환을 가진 산모에게서 태어난 아기는 △선천성 심장질환 △얼굴 기형 △저체중아 출산 △작은 머리 크기(소두증) 등의 증상을 가질 수 있다. 반면 PKU가 잘 관리되면 건강한 아기를 가질 수 있다.
페닐케톤뇨증은 PAH 유전자 이상으로 PAH 효소가 결핍되거나 소실돼 유발된다. PAH 효소가 없으면 페닐알라닌을 분해할 수 없기 때문에 몸에 축적되고 결국 증상을 야기한다.
페닐알라닌은 필수아미노산으로서 인체에 필요하지만 자연적으로 생성되지 않는 아미노산이다. 그래서 음식을 통해 섭취해야 한다. 육류, 계란, 유제품 등 고단백 식품에 풍부하다. 일반적으로 인체는 필요한 페닐알라닌을 사용하고, 남은 페닐알라닌은 다른 물질로 전환시킨다. 하지만 아기에게 PAH가 부족하면 페닐알라닌이 혈액과 뇌에 축적된다.
진단
페닐케톤뇨증은 상염색체 열성 유전으로 유전된다. 따라서 아기가 양쪽 부모로부터 PAH 변이 유전자를 각각 받아야 이 질환이 발병한다.
혈액 몇 방울이면 쉽게 페닐케톤뇨증을 진단할 수 있다. 생후 2~3일경 된 신생아의 발뒤꿈치에서 혈액을 채취하여 신속한 선별검사(Guthrie 법)로 진단한다. 혈액에서 페닐알라닌 수치가 높으면 추가 검사나 소변검사를 한다. 페닐케톤뇨증은 소변을 이용한 염화제이철 반응에서 녹색을 보인다.
혈중 페닐알라닌이 지속적으로 4mg/dL 이상이면 비정상이다. 지속적으로 20mg/dL 이상이면 전형적(typical, 또는 Classical) 페닐케논뇨증, 4~20mg/dL인 경우는 고페닐알라닌혈증으로 구분한다. 후자는 치료하지 않아도 지능이 정상이거나 장애 정도가 가볍다.
전형적 페닐케톤뇨증으로 판명되면, 페닐알라닌수산화효소에 대한 검사나 유전자검사를 시행해 확진한다. 이 때 BH4 반응성 검사를 통해 비전형적(atypical) 페닐케톤뇨증인지 재차 확인하게 된다. 페닐알라닌을 티로신으로 전환하는 PAH 효소의 필수 보조인자가 BH4(tetrahydrobiopterin, 또는 THB)이다. 전체 PKU의 약 2%가 BH4 부족(대사결핍)으로 일어나며 이는 비전형적 PKU로 분류된다. BH4 생합성에 관련된 효소에 대한 검사나 유전자검사를 시행함으로써 비전형적 여부를 확정하게 된다.
산모가 임신 중이라면 산전 유전자검사를 통해 아기가 페닐케톤뇨증을 포함한 유전질환에 걸릴 위험이 있는지 확인할 수 있다. 이미 PKU를 가진 아이를 출산했다면 산전 유전자검사가 긴요하다. 같은 질환을 가진 아이를 다시 임신할 위험이 더 높기 때문이다.
PKU는 완치할 수 없다. 하지만 치료를 통해 증상과 심각한 합병증 발생을 예방, 관리할 수 있다. 페닐케톤뇨증 치료는 평생 지속되며, 특별한 식이요법과 약물 복용으로 관리한다. 만약 이런 치료를 게을리하면 증상이 빠르게 재발한다.
페닐알라닌 다이어트
PKU 환자는 페닐알라닌 함량이 낮은 특별 식단을 준수해야 한다. 고기, 계란, 유제품처럼 단백질 함량이 높은 식품에는 페닐알라닌이 다량 함유돼 있다. 신체활동에 어느 정도의 단백질이 필요하므로 영양사의 도움을 받아 균형잡인 식단을 영위토록 신경써야 한다.
인공감미료인 아스파탐도 피해야 한다. 아스파탐은 체내에서 소화될 때 페닐알라닌을 혈류로 방출하기 때문이다. 다이어트식품 또는 무설탕‧무가당 식품 또는 의약품에 아스파탐이 종종 들어가므로 유의해야 한다. 예컨대 시리얼 제품, 저당 요구르트, 다이어트 소다, 가향 탄산수, 무설탕 껌, 저칼로리 감미료, 씹어먹는 비타민, 기침약 등에 아스파탐이 자주 첨가된다.
페닐케톤뇨증이 있고 임신 중이라면, 임신 기간에 이 식단을 철저히 준수해야 한다. 이렇게 하면 아기가 건강하게 태어날 수 있다. 페닐케톤뇨증이 있는 신생아는 출생 즉시 페닐알라닌이 없는 특수 분유를 섭취해야 한다.
전형적 페닐케톤뇨증으로 진단되면 즉시 저 페닐알라닌 특수분유를 먹이고 BH4 반응성을 보이는 경우는 BH4(≤20mg/kg/day)를 투여한다. 여기서 BH4는 바이오마린의 ‘쿠반’을 지칭한다(후술). 6세 이전까지는 저 페닐알라닌 식단을 철저히 유지하고, 성장기에 본격 접어드는 식단을 다소 완화해도 된다. 사춘기부터 평생 동안 혈중 페닐알라닌치를 3~15 mg/dL로 유지하는 것이 바람직하다, 페닐알라닌 섭취량이 필요량보다 지나치게 적으면 발육장애, 빈혈, 저단백혈증 등 페닐알라닌 결핍 증상이 나타난다. 하지만 어떤 경우에도 식이요법을 중단하는 것은 절대적으로 금물이다.
PKU 치료 약물
미국 식품의약국(FDA)은 페닐케톤뇨증의 치료제로 3가지 약물을 승인했다.
바이오마린의 ‘쿠반’(Kuvan, 성분명 Sapropterin dihydrochloride)
바이오마린이 2007년 12월 13일에 승인받은 약이다. 쿠반(사프로프테린 이염산염, 또는 사프롭테린)은 페닐알라닌 수산화효소와 함께 작용하여 페닐알라닌(Phe)을 대사하는 천연 효소 보조인자인 6R-BH4(테트라히드로비옵테린)의 합성 물질이다. 전형적 또는 비전형적 PKU에 쓸 수 있으나 ‘sapropterin-responsive’ 유형의 PKU여야 효과를 볼 수 있다. 다시 말해 사프로프테린 투여로 PAH의 기능 활성화가 관찰되는 경우에만 쓸 수 있다. BH4 결핍이 일어난 비전형적 PKU에 더욱 효과적인 것이다. 정제로서 반드시 페닐케톤뇨증 식이요법을 병행해야 한다.
등록 환자를 대상으로 한 ‘PKUDOS’ 장기 연장 임상시험 결과 혈중 페닐알라닌 농도를 591 µmol/L에서 392 µmol/L로 34% 감소시키는 것으로 나타났다. 다른 임상에서는 6주 만에 이 수치를 236 µmol/L 낮췄다. 반면 위약은 오히려 3 µmol/L 증가했다. 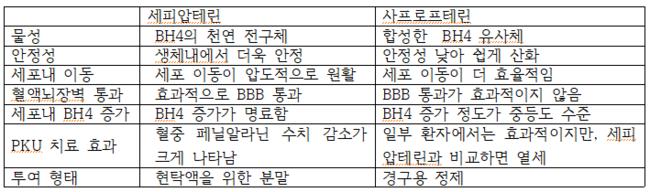
PTC테라퓨틱스의 ‘세피언스’(Sephience, 성분명 sepiapterin)
PTC테라퓨틱스가 2025년 7월 28일에 승인받은 약이다. 세피언스는 세피압테린(또는 세피아프테린) 반응성 페닐케톤뇨증(sepiapterin-responsive phenylketonuria)을 앓는, 고페닐알라닌혈증(hyperphenylalaninemia) 환자(생후 1개월 이상 소아 및 성인)의 치료제로 광범위한 적응증을 허가받았다.
세피언스는 PAH의 중요한 효소 보조인자인 BH4의 자연 전구체다. PAH를 활성화시키는 작용 기전을 통해 혈중 페닐알라닌(Phe) 수치를 효과적으로 낮출 수 있고 광범위한 페닐케톤뇨증 환자에게 치료 가능성을 제공한다.
세피언스는 3상 ‘APHENITY’ 임상시험에서 입증된 유의한 유효성 및 안전성과 APHENITY 장기 연장 연구에서 확인된 치료 효과의 지속성을 근거로 승인이 이뤄졌다.
임상시험 결과 세피언스는 전체 치료 대상 집단에서 페닐알라닌 수치를 평균 63% 감소시켰고 전형적 페닐케톤뇨증 환자 하위집단에서는 69% 감소시킨 것으로 나타났다. 피험자의 대다수(84%)는 치료 가이드라인에서 권고되는 페닐알라닌 수치인 360 µmol/L 이하를 달성했고, 피험자의 22%는 페닐알라닌 수치가 정상 범위에 도달했다.
페닐알라닌 내성 하위그룹 분석에서는 피험자의 약 60%가 페닐알라닌 수치를 360 µmol/L 미만으로 유지하면서 연령별 권장 단백질 섭취량을 초과하는 수준의 단백질 섭취가 가능한 것으로 관찰됐다. 이러한 결과는 세피언스가 페닐알라닌 수치 조절을 유지하면서 환자들이 따라야 하는 매우 제한적인 식이요법을 완화할 수 있음을 뒷받침한다.
세피압테린은 BH4의 천연 전구체로서, 합성 BH4 유사체인 사프로프테린에 비해 더 안정적이고 약효과 우수하다. 세피압테린은 세포 내 BH4로 더 효율적으로 전환되고, 혈액-뇌 장벽을 통과한다. 또 임상 연구에서 사프로프테린보다 페닐알라닌 수치 감소에 더 우수한 효능을 보였다. 사프로프테린은 세포 내 BH4 수치를 중등도로 증가시키는 반면, 세피압테린은 현저하게 증가시킨다.
바이오마린의 ‘팰린직’(Palynzig, 성분명 pegvaliase-pqpz)
팰린직은 2018년 5월 24일, 성인 페닐케톤뇨증 환자용으로 허가를 취득한 최초이자 유일한 효소 대체요법제이다. 기존 약물치료 및 저단백 식이요법에도 불구하고 페닐알라닌 수치가 조절되지 않은 경우에 투여한다.
이 약에는 식물 원료(Anabaena variabilis, 사상형 남조류, filamentous cyanobacterium)에서 유래한 효소 페닐알라닌 암모니아 분해효소(phenylalanine ammonia-lyase, PAL)의 PEGylated(폴리에틸렌 글리콜 변형) 버전 활성 물질인 페기발리아제(pegvaliase)가 함유돼 있다.
이런 대체 PAL 효소는 페닐알라닌 분해를 위한 대체 경로를 제공하여 페닐알라닌(Phe)을 암모니아와 트랜스-시나민산(trans-cinnamic acid)이라는 두 가지 분자로 전환된다. 이렇게 전환된 대사산물은 간에서 대사되어 결국 체외로 배출된다. 이로써 과잉의 페닐알라닌을 효과적으로 대사시켜 안전한 혈액 Phe 수치로 낮춰주고 높은 Phe 농도와 관련된 심각한 건강 문제를 해소한다.
혈중 페닐알라닌 수치가 제대로 관리되지 않는 일부 성인에서 도움이 된다. 피하주사제로서 페닐알라닌 분해효소를 대체할 수 있다. 팰린직은 성인의 단백질 섭취량을 늘리거나 정상적인 식단을 유지할 수 있도록 도울 수 있다. 하지만 단백질을 늘려도 되는지는 의사와 지속적으로 상의해야 한다.
팰린직은 2025년 7월 29일, 기존 18세 이상 성인에서 12~17세 청소년으로 적용 연령대를 넓히기 위한 추가 신청서가 FDA에 제출됐다. 2026년 2월 28일 이전에 승인 여부가 결정될 예정이다.
팰린직은 24개월 이상 투여한 86명 중 57명(66%)에서 페닐알라닌 농도가 치료 가이드라인에서 권고되는 페닐알라닌 수치인 360 µmol/L 이하를 달성했다. 또 36개월 이상 투여한 77명 중 51명(66%)이 페닐알라닌 수치인 360 µmol/L 이하에 도달했다. 118명 중 89명(75%)가 짧게는 2주 만에, 길게는 57개월 만에 이 목표에 도달했다. 평균 달성 소요 기간은 13개월이었다.
24개월 이상 투여한 환자 중 120 µmol/L를 달성한 비율은 86명 중 43명으로 50%였다. 36개월 이상 투여한 환자 중 120 µmol/L를 달성한 비율은 77명 중 37명으로 48%였다. 이같은 임상 결과는 일반 PKU 환자로서 이전에 다른 약물을 복용해 치료에 실패한 환자가 아니다.
이 약은 중대한 이상반응으로 아나필락시스가 올 수 있어 유의해야 한다. 가장 흔한 이상반응으로는 주사 부위 반응, 관절통, 과민반응, 두통, 최소 14일 이상 지속되는 전신성 피부 반응, 가려움증, 메스꺼움, 현기증, 복통, 인후통, 피로, 구토, 기침, 설사 등이다. 과민반응은 대부분의 환자에서 발생했는데 이 약에 대한 항체 형성 때문인 것으로 분석됐다.
한국에서 허가된 페닐케톤뇨증 치료제로는 GC녹십자의 디테린정(성분명: 사프로프테린 이염산염, 2025년 희귀의약품 지정, 2017년 2월 국내 출시)과 2025년 7월 3일 희귀의약품으로 지정된 ‘세피언스’(성분명: 세피압테린)가 있다. 팰린직은 국내에 허가되지 않았다.
현재 개발 중인 신약후보물질
일본 오츠카제약에 인수된 2024년 8월 1일에 인수된 미국 보스턴의 냐나테라퓨틱스(Jnana Therapeutics)는 ‘JNT-517’을 개발 중이다. 이 신약후보는 소장과 신장에서 아미노산 재흡수를 조정하는 아미노산 수송체 ‘SLC6A19’의 활동을 저해하는 알로스테릭 억제제다. 신장을 통해 페닐알라닌을 배출을 유도할 것으로 예상되고 있다. 현재 성인 PKU 환자를 대상으로 3상 임상이 진행 중이다. 120명 중 3분의 2가 JNT-517를, 나머지 3분의 1이 위약을 투여받게 된다.
미국 캘리포니아주 월넛크리크(WALNUT CREEK) 소재 넥스트제네레이션진테라퓨틱스(Next Generation Gene Therapeutics, NGGT)가 유전자치료제 ‘NGGT002’로 1/2상을 진행 중이다. 2024년 12월에 발표된 중간 연구결과 성인 전형적 PKU 환자 6명 중 5명은 이 약을 투여받은 뒤 3주 후 혈장 페닐알라닌 수치가 정상 범위 내로 유지됐다. 앞서 연구자주도임상(Investigator Initiated Trial. IIT)에서 NGGT002를 처음 투여받은 환자는 약물 투여 40주 후에도 혈장 페닐알라닌 수치가 정상 범위 내로 유지됐다.
아메리칸진테크놀로지(American Gene Technologies)와 바이오마린도 유전자치료제 개발을 위한 연구를 진행하고 있다. 유전자치료는 단회 치료로 완치되거나, 한번 치료 후 효과가 장기간 유지되도록 하기 위해 기획됐다. 주로 CRISPR 유전자편집기술로 결핍된 유전자를 정상 유전자로 교정하는 방식이다.
조언
페닐케톤뇨증은 평생 지속되는 질환이기 때문에 조기 진단을 통해 심각한 합병증이 일어나지 않도록 증상을 관리하고 건강에 미치는 영향을 최소화해야 한다. 평생 저단백 식단을 유지해야 하는 경우가 대부분이어서 영양사나 의사 등으로부터 도움을 받아야 한다. 환자단체(자조집단)에 가입하는 것도 좋은 방법이다.
처음 진단을 받아 충격에 빠지거나, 평생 치료해야 한다는 사실이 좌절스럽다면 정신 건강 전문가에게 도움을 요청해야 한다. 가족력이 있어 고위험군이라면 유전학 전문의와 상의해 보다 정밀한 관리를 받아야 한다.

 목록
목록

























